top of page

SCIENCE & PIPELINE
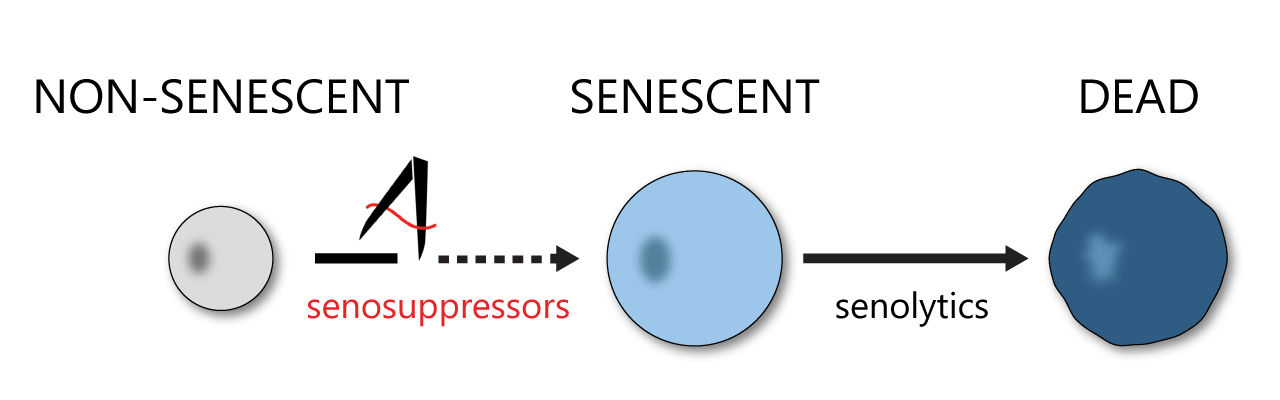
Atropos is a drug discovery platform to identify chemical matter that can modulate the formation of senescent cells, both for aging applications (senosuppressors) and oncology applications (senostimulators).
What are senescent cells?
Senescence is classically thought of as a cellular state induced by various stressors in which cells irreversibly exit the proliferative cycle and elaborate a program of gene expression and secretion of cytokines (SASP) that leads to inflammation. These cells are also largely resistant to physiological death signals. Senescence can be beneficial, in the case of wound healing and cancer therapy, or harmful in the case of aging.
A revolution in our understanding of the contribution that cellular senescence makes to organismal aging was ushered in by the seminal studies of van Duersen and Campisi, who showed that the accumulation of senescent cells in mice induced inflammation associated tissue damage and the decline of organismal fitness, ie aging.
The van Duersen and Campisi work suggested that removing accumulated senescent cells will improve healthspan (the healthy period of one’s life) by preventing degeneration of, or damage to, vital organs and tissues. This gave rise to a new industry aimed at creating senolytic drugs, or drugs that specifically and selectively eliminate such cells. However, there is a lack of understanding of how senescent cells accumulate in an aging organism or what the specific targets are in the various tissue or organ systems that make them different from normal cells. This makes it difficult to selectively target such cells and has hampered the development of senolytic drug.


Our platform
FATES (First ATRX-based Therapeutics to Elucidate Senescence, patent pending) is a cell-based phenotypic screening platform based on the detection of de novo ATRX foci. It is a rapid and quantitative drug discovery platform. It has been scaled and piloted with Scripps Molecular Medicine in Florida and has identified chemical matter that allows manipulation of SAGA for applications in aging (senosuppressors). This platform technology can also be used to identify novel anti-cancer drugs (senostimulators).
Our approach
Atropos capitalizes on a revolutionary new understanding of cellular senescence obtained when our scientific founder discovered that cells can progress directly from quiescence, or reversible cell cycle exit, to senescence (SAGA; Senescence After Growth Arrest). Understanding the molecular mechanism and genetic requirements of this transition opens up the possibility that one can target the rate of formation of such cells without affecting inflammation or promoting cell proliferation. Defining this pathway was originally based on therapy induced senescence in cancer cells treated with cdk4/6 inhibitors. Work in Atropos laboratories has expanded upon this and demonstrated that some of the original molecular and genetic events identified during therapy induced senescence are in fact conserved, and necessary, including in systems associated with aging in primary cells.
Targeting the formation of senescent cells is not a “new idea”. Others have approached this, and continue to do so, either by manipulating signaling pathways such as TOR or more directly manipulating cellular metabolism by altering NAD levels. Atropos has identified molecular events that are unique to SAGA, a notable advantage that holds the promise of selective and specific regulation of cellular senescence.
Selected literature
Baker DJ, Wijshake T, Tchkonia T, LeBrasseur NK, Childs BG, van de Sluis B, Kirkland JL, van Deursen JM. Clearance of p16Ink4a-positive senescent cells delays ageing-associated disorders. Nature (2011). PMID: 22048312.
Chang J, Wang Y, Shao L, Laberge RM, Demaria M, Campisi J, Janakiraman K, Sharpless NE, Ding S, Feng W, Luo Y, Wang X, Aykin-Burns N, Krager K, Ponnappan U, Hauer-Jensen M, Meng A, Zhou D. Clearance of senescent cells by ABT263 rejuvenates aged hematopoietic stem cells in mice. Nat Med (2016). PMID: 26657143.
Kovatcheva M, Liu DD, Dickson MA, Klein ME, O'Connor R, Wilder FO, Socci ND, Tap WD, Schwartz GK, Singer S, Crago AM, Koff A. MDM2 turnover and expression of ATRX determine the choice between quiescence and senescence in response to CDK4 inhibition. Oncotarget (2015). PMID: 25803170.
Kovatcheva M, Liao W, Klein ME, Robine N, Geiger H, Crago AM, Dickson MA, Tap WD, Singer S, Koff A. ATRX is a regulator of therapy induced senescence in human cells. Nat Commun (2017). PMID: 28855512.
Kovatcheva M, Klein ME, Tap WD, Koff A. Mechanistic understanding of the role of ATRX in senescence provides new insight for combinatorial therapies with CDK4 inhibitors. Mol Cell Oncol (2017). PMID: 29404388.
Klein ME, Dickson MA, Antonescu C, Qin LX, Dooley SJ, Barlas A, Manova K, Schwartz GK, Crago AM, Singer S, Koff A, Tap WD. PDLIM7 and CDH18 regulate the turnover of MDM2 during CDK4/6 inhibitor therapy-induced senescence. Oncogene (2018). PMID: 29789718.
Klein ME, Kovatcheva M, Davis LE, Tap WD, Koff A. CDK4/6 Inhibitors: The Mechanism of Action May Not Be as Simple as Once Thought. Cancer Cell (2018). PMID: 29731395.
bottom of page